Аннотация
В работе описываются наиболее важные параметры, относящиеся к математическим моделям
вторичных и третичных структур молекул рибонуклеиновых кислот (РНК). Дается строгая
количественная оценка разнообразия возможных вторичных структур молекул РНК. Ставится задача
идентификации параметров модели третичной структуры, основывающаяся на данных о третичных
структурах реальных молекул транспортных РНК, полученных методом рентгеноструктурного анализа.
Первый этап этой задачи (определение геометрических параметров двуспиральных участков
третичной структуры молекулы РНК) решается на основе рентгеноструктурных данных о форме
дрожжевой фенилаланиновой транспортной РНК.
Abstract
The most important parameters of mathematical models of secondary and tertiary structures
of molecules of ribonucleic acids (RNA) are described in this work. A severe quantitative
estimation of a variety of possible secondary structures of RNA molecules is given. A task
of identification of the tertiary structure model's parameters based on the structures of
real transport RNAs received by a method of the X-Ray analysis is put. The first stage of
this task (determination of geometrical parameters of two-spiral sites of tertiary structure
of an RNA molecule) is solved on the basis of the X-Ray data on the form of a yeast
phenilalanine transport RNA.
Содержание
Введение.............................................................................................................. 3
1. Разнообразие структур молекул РНК......................................................... 5
1.1 Иерархия структур нуклеиновых кислот................................................. 5
1.2 Элементы пространственных структур РНК........................................... 6
1.3 Типы петель во вторичной структуре...................................................... 7
1.4 Способы описания вторичной структуры............................................... 9
1.5 Стерическое условие и псевдоузлы....................................................... 10
1.6 Оценка
количества вторичных структур РНК....................................... 10
1.7 Разнообразие
вторичных структур для тРНК....................................... 13
2. О
задаче идентификации параметров....................................................... 13
2.1 Математическая
модель пространственной структуры РНК................ 13
2.2 Система
уравнений модели.................................................................... 14
2.3 Параметры
модели.................................................................................. 15
2.4 Постановка
задачи идентификации........................................................ 15
2.5 Генетический
алгоритм поиска экстремумов........................................ 16
2.6 Задача
сравнения двух ломаных............................................................ 17
2.7 Определение
параметров Уотсон-Криковской связи............................ 22
3. Результаты
вычислительных экспериментов........................................... 26
Литература......................................................................................................... 28
Математическое моделирование
пространственной структуры биологических макромолекул, таких как РНК и белки,
является в настоящее время одной из наиболее бурно развивающейся ветвей
молекулярной биологии. Фундаментальность этой проблемы определяется тем, что
основные процессы жизнедеятельности клетки зависят в первую очередь от
пространственной структуры упомянутых макромолекул.
В последнее время в
фармакологической промышленности интенсивно развивается метод создания
лекарственных препаратов на основе РНК-содержащих соединений (SELEX, Systematic Evolution of Ligands by Exponential enrichment [11, 12]). Он основан на том, что даже сравнительно короткие молекулы РНК
(около 100 нуклеотидов) обладают гигантским количеством пространственных форм.
Такое разнообразие в принципе позволяет подобрать подходящую по форме молекулу
РНК, закрывающую активные центры патогенного соединения (белка или фермента) и
препятствующую его разрушительной работе (docking-метод [8]). Основная проблема здесь – это поиск достаточно дешевого метода определения
пространственной структуры короткой молекулы РНК по ее нуклеотидному составу
(первичной структуре). Наиболее перспективными для решения этой задачи мы
считаем методы математического моделирования.
Для определения пространственной структуры
молекулы чаще всего используется рентгеноструктурный анализ. Однако, этот
физический метод является дорогостоящим и длительным. Помимо чисто практических
нужд фармакологии, необходимость в дешевых методах определения третичной
структуры молекул РНК вызывается и тем, что поток информации о вновь открытых молекулах
и расшифровке их нуклеотидного (для нуклеиновых кислот) и аминокислотного (для
белков) состава год от года возрастает.
Предлагаемая нами математическая модель
пространственной структуры РНК [1] является довольно простой, и вряд ли она
будет давать точную информацию о пространственной структуре длинных молекул
РНК. Тем не менее, при определении пространственной структуры другими методами
нашу модель можно использовать для построения начального приближения структуры.
Затем ее можно последовательно уточнять.
Молекулярная цепь рассматривается как тонкий
упругий стержень, имеющий в свободном состоянии форму винтовой линии. С шагом,
равным длине одного нуклеотида, в стержень вделаны жесткие перемычки, равные по
длине половине Уотсон-Криковской связи. В простейшей модели параметры винтовой
линии и ориентация перемычек выбираются так, что два свободных стержня
одинаковой длины, будучи правильно расположены, образуют двойную спираль в
A-форме. Пространственная структура молекулы собирается из стеблей и петель в
соответствии с заданной вторичной структурой.
Каждая петля состоит из семейства тонких упругих стержней со взаимно
согласованными краевыми условиями равновесия. В соответствии с краевыми
условиями концы стержней ориентируются так же, как концы нитей в стебле А-формы
РНК.
Качество предсказания
пространственной структуры молекулы РНК напрямую зависит от правильного
определения параметров математической модели. В данной работе рассматриваются
вопросы, связанные с решением этой
задачи.
Параметры модели разбиваются на несколько
групп. Это геометрические параметры стержня, моделирующего молекулярную цепь,
его упругие характеристики, геометрические параметры жестких перемычек,
моделирующих Уотсон-Криковскую связь, и геометрические параметры двуспиральных
участков.
Задача идентификации параметров модели
естественным образом разделяется на два этапа. Первый этап – это идентификация
геометрических параметров двуспиральных участков и жестких перемычек,
моделирующих Уотсон-Криковскую связь.
Второй этап – это идентификация геометрических и упругих параметров
стержня, моделирующего молекулярную цепь,
с использованием результатов, полученных на первом этапе.
В работе основное внимание уделяется
первому этапу задачи идентификации. Основная идея идентификации параметров
состоит в использовании опубликованной информации о третичных структурах
некоторых реальных молекул транспортных РНК, полученной методом
рентгеноструктурного анализа [10]. Эти данные включают, в частности, координаты
атомов рибозофосфатных остовов обеих нитей, составляющих двуспиральные участки
стеблей тРНК. Идентифицируемые параметры подбираются таким образом, чтобы нити
модельного двуспирального участка как можно лучше приближались к нитям рибозофосфатных остовов двуспирального
участка реальной молекулы.
Изложим теперь кратко содержание
препринта. Для большей ясности, помимо материала, посвященного собственно
задаче идентификации параметров, в работу включен раздел, в котором кратко
описываются основные понятия, задачи и параметры, относящиеся к общей проблеме
изучения пространственных структур молекул РНК. В этом же разделе дается
строгая количественная оценка разнообразия структур молекул РНК, которая
показывает перспективность использования РНК-содержащих препаратов в
фармакологии.
Материал, относящийся к задаче
идентификации параметров, разбит на три раздела. Первый раздел посвящен оценке
разнообразия потенциально возможных структур молекул РНК. Во втором разделе
дается строгая формулировка задачи идентификации, рассматриваются проблемы,
возникающие при ее решении. Также описываются методы и алгоритмы разрешения
этих проблем. Третий раздел посвящен описанию результатов первых экспериментов
по определению геометрических параметров двуспиральных участков третичной
структуры молекулы РНК на основе данных рентгеноструктурного анализа формы
дрожжевой фенилаланиновой транспортной РНК [10]. Опишем содержание работы более подробно.
В первом разделе дается
краткое описание иерархии структур нуклеиновых кислот и, в частности, структур
молекул РНК. Принято выделять четыре структурных уровня – первичную, вторичную, третичную и четвертичную структуры молекулы
РНК.
В рассматриваемой модели пространственная
(третичная) структура строится на основе известной вторичной. Краткий обзор по
вторичным структурам можно найти, например, в [6, 7]. Задача об определении
вторичной структуры в данной работе не рассматривается. Однако, здесь дается
оценка числа потенциально возможного количества вторичных структур.
Оказывается, что количество вторичных структур, имеющих n связей, пропорционально 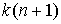 . Символом k() мы обозначаем так называемые числа
Каталана, изучаемые в комбинаторике [3]. . Символом k() мы обозначаем так называемые числа
Каталана, изучаемые в комбинаторике [3].
Во втором разделе дается краткое описание
используемой математической модели и ее параметров, подлежащих идентификации.
Рассказывается о постановке задачи идентификации, дается описание данных
рентгеноструктурного анализа молекул тРНК, называются основные шаги процесса
идентификации. Далее описывается генетический алгоритм поиска экстремумов и способ
построения хромосомы для задачи идентификации. Также рассматривается задача
сравнения двух ломаных с различным числом звеньев. Предлагаются три методики ее
решения.
В третьем разделе приводятся результаты
некоторых вычислительных экспериментов по оценке геометрических параметров
модели двуспиральных участков РНК на основе рентгеноструктурных данных. B задаче сопоставления геометрических форм
пространственных кривых функционал сравнения может иметь не одну точку
экстремума. Следует ожидать, что их может быть довольно много. Поэтому в
строгом математическом смысле задача не является корректной (т. е. ее решение
не единственно). Однако, наличие минимума у оценочного функционала можно
гарантировать, т. к. он ограничен снизу на компактном пространстве параметров.
Поэтому осмысленной является задача изменения геометрических параметров модели
таким образом, чтобы оценочный функционал стал меньше при разумной форме и
взаимном расположении сопоставляемых кривых в точке экстремума. В силу
сказанного результаты процесса идентификации контролировались нами для того,
чтобы отбрасывать заведомо неподходящие варианты. В разделе приводятся
результаты наиболее удачных процессов идентификации параметров.
В
данном разделе рассматриваются вопросы,
связанные с оценкой разнообразия потенциально возможных структур молекул РНК.
Химически молекула РНК представляет собой
полинуклеотид – цепочку соединенных в линейной последовательности мономеров-нуклеотидов.
В РНК нуклеотиды бывают 4х типов: аденин (A), гуанин (G), цитозин (C) и урацил (U). Последовательность расположения
нуклеотидов в молекулярной цепочке несет информационную нагрузку: это могут
быть копии генов, необходимых клетке для процесса жизнедеятельности (матричная
РНК). В иных случаях последовательность нуклеотидов задает форму молекулы,
определяющую функцию РНК (транспортные РНК, рибосомальные РНК). Нуклеотидная
последовательность молекулы называется ее первичной
структурой.
Некоторые нуклеотиды в молекулярной
цепочке связаны попарно т.н. Уотсон-Криковскими связями. Структура этих связей
называется вторичной структурой РНК.
В силу конечности множества возможных Уотсон-Криковских связей число вторичных
структур, которые может принимать данная молекула РНК, конечно, но весьма
значительно. Определение реальной вторичной структуры РНК по ее известной
первичной структуре также является важной фундаментальной проблемой
молекулярной биологии.
Под третичной
структурой молекулы РНК (ДНК) понимается пространственная форма, которую
принимает ее молекулярная цепочка в пространстве под воздействием
Уотсон-Криковских и других более слабых потенциалов. Четвертичной структурой называется
форма молекулы, которую она приобретает, связываясь в комплекс с другими
биомолекулами. Первичная структура у молекулы РНК одна, а возможных вторичных
(третичных, четвертичных) структур много.
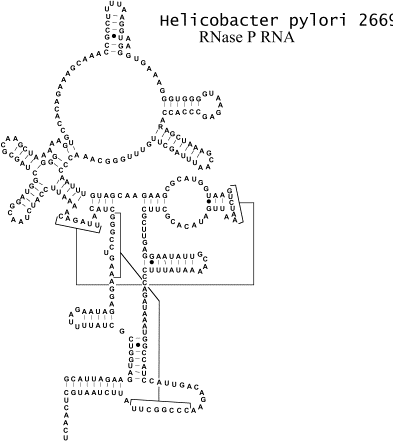
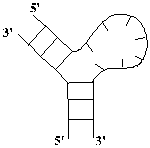
Рис. 1.1
Первичная и вторичная структуры молекулы Helicobaster pyori RNase P RNA [9].
С точки зрения вторичной структуры все
нуклеотиды в молекуле можно разбить на два класса – спаренные (т.е. образующие
Уотсон-Криковскую связь с каким-либо другим нуклеотидом) и свободные
(неспаренные). Формально, вторичная
структура РНК – это описание всех спаренных и свободных оснований в
молекулярной цепи.
Отрезок молекулярной цепи, состоящий из
неспаренных оснований, называется однонитевым. Отрезки [n1, n2] и [n3, n4] спаренных оснований так, что n1 спарен с n4, n1 +1 с n4 -1,…, n2 с n3, образуют двухнитевый или двуспиральный участок во вторичной структуре РНК.
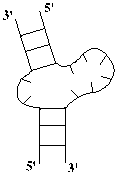
Однонитевые участки (и семейства таких участков) называются петлями, а двухнитевые – стеблями. Стебель можно представить
себе, как участок винтовой лестницы, где ступеньки – это поперечные, Уотсон-Криковские
связи. Длиной стебля называется число пар оснований в нём: n2 - n1 + 1 = n4 - n3 + 1. Таким образом, вторичная структура РНК – это
совокупность стеблей и петель.
Рис. 1.2
Однонитевый (петля) и двухнитевый (стебель) элементы вторичной структуры РНК.
Петлей называется замкнутая последовательность
однонитевых участков РНК, концы которых соединены Уотсон-Криковскими вторичными
связями. При этом начало каждого следующего участка соединено с концом предыдущего,
а конец последнего участка соединен с началом первого. Однонитевые участки,
входящие в состав петли, называются ее ветвями или звеньями. Длиной петли называется число свободных
нуклеотидов, входящих в ее состав. Выделяют
следующие типы петель.
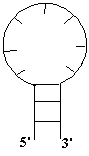 
a) Шпилечная
петля b) Боковая петля
 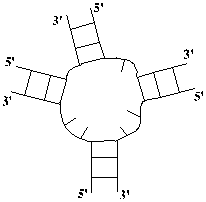
c) Внутренняя
петля d) Многозвенная петля
Рис 1.3 Типы
петель (однонитевых участков вторичной структуры).
Шпилечная
петля соединяет первую и
вторую нить в одном стебле. Это простейшая однозвенная петля (она состоит из
одного однонитевого участка). Считается, что шпилечная петля всегда содержит не
менее трех нуклеотидов.
Боковая
петля содержит два однонитевых
участка, один из которых вырожден – имеет нулевую длину (не содержит ни одного
несвязанного нуклеотида). Длина же второго участка называется длиной боковой
петли.
Внутренняя
петля содержит два
однонитевых участка. Длины этих участков
являются параметрами, определяющими петлю.
Многозвенная
петля содержит несколько
однонитевых участков. Число этих участков и их длины являются параметрами,
определяющими петлю.
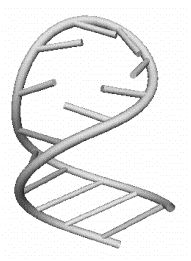
a) b)
Рис 1.4 Элементы третичной структуры РНК: a) стебель; b) шпилечная петля длиной в семь нуклеотидов.
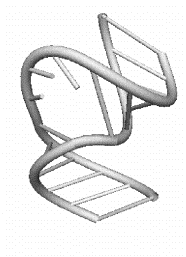
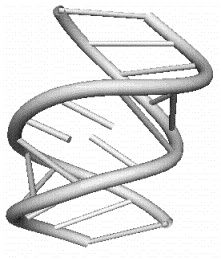
a)
b)
Рис 1.5 Элементы третичной структуры РНК: a) боковая петля, состоящая из трех
нуклеотидов; b) внутренняя
петля, звенья которой состоят из одного и двух нуклеотидов.
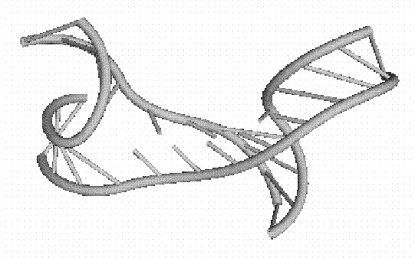
Рис 1.6 Элементы
третичной структуры РНК: пример многозвенной петли, состоящей из четырех
звеньев.
Существует несколько способов описания
вторичных структур РНК. Мы опишем два основных.
Геометрический
способ – молекулярная цепь располагается на плоскости так, чтобы стебли
образовывали прямоугольные “лесенки”, а петли – замкнутые контуры (см. Рис 1.2,
1.3). Такой способ дает некоторое представление о реальной геометрии структуры
молекулы.
Представление
на окружности – молекулярная цепь располагается на плоскости по окружности, так что
движению от 5’ конца к 3’
концу соответствует движение против часовой стрелки. Уотсон-Криковские
связи при этом изображаются хордами
окружности. Если никакие две хорды не пересекаются, то говорят, что
выполняется стерическое условие.
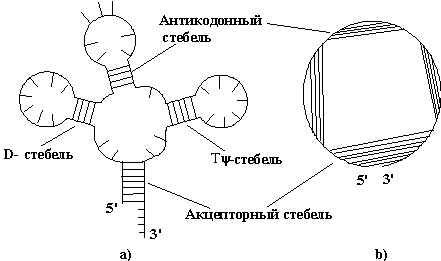
Рис 1.7
Представление вторичной структуры тРНК геометрически (a) и на окружности (b). Для такой структуры стерическое условие
выполнено.
Если в представлении вторичной структуры
на окружности никакие две хорды не пересекаются, то говорят, что выполняется стерическое условие. Перенумеруем все
нуклеотиды в молекулярной цепи, начиная с 1.
Две пары нуклеотидов (p1, p2) и (q1, q2) стерически совместимы, если их связи не “перекрещиваются”, т.е. выполнено
одно из условий:
либо [p1, p2] Í
[q1, q2], либо [q1, q2] Í
[p1, p2], либо [p1, p2] Ç [q1,
q2] = Æ,
где [n, m] – это целочисленный отрезок от n до m. Два стебля стерически совместимы, если
совместимы любые две составляющие их Уотсон-Криковские пары. Если стерическое условие не выполнено, то
вторичная структура содержит псевдоузлы. В описываемой модели предполагается, что
псевдоузлы во вторичной структуре отсутствуют.
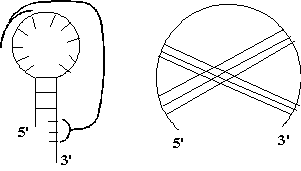
Рис. 1.8
Псевдоузел. Связи, входящие в него, пересекаются на круговой диаграмме, т. е.
нарушается стерическое условие.
В подавляющем большинстве случаев
различные вторичные структуры соответствуют различным третичным. Поэтому оценка
количества вторичных структур дает представление о количестве третичных
структур. Попробуем оценить количество вторичных структур, которые могут
возникать в молекулярной цепи, первичная структура которой (т.е. нуклеотидный
состав) случайна.
Будем называть длиной структуры количество Уотсон-Криковских связей в ней.
Напомним, что мы рассматриваем только структуры, удовлетворяющие стерическому
условию. Обозначим за 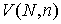 среднее число
структур длины среднее число
структур длины 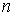 при случайном выборе молекулярной цепи, состоящей из при случайном выборе молекулярной цепи, состоящей из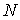 нуклеотидов. Количество структур длины 1 равно числу стеблей, содержащих только
одну Уотсон-Криковскую связь: нуклеотидов. Количество структур длины 1 равно числу стеблей, содержащих только
одну Уотсон-Криковскую связь:
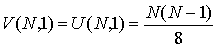 (1.1) (1.1)
Рассмотрим теперь структуру длины n. В ней какие-то 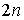 нуклеотидов попарно
связаны Уотсон-Криковскими связями. Всего возможно нуклеотидов попарно
связаны Уотсон-Криковскими связями. Всего возможно 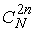 различных выборов различных выборов 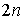 нуклеотидов. После того, как нуклеотиды выбраны, вторичная
структура определяется системой связей между ними. Перенумеруем выбранные
нуклеотиды последовательно от 1 до нуклеотидов. После того, как нуклеотиды выбраны, вторичная
структура определяется системой связей между ними. Перенумеруем выбранные
нуклеотиды последовательно от 1 до 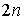 и расположим их в
вершинах правильного и расположим их в
вершинах правильного 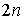 -угольника. Соединим связанные нуклеотиды диагоналями. Каждой вторичной структуре взаимно
однозначно соответствует набор из n непересекающихся диагоналей. -угольника. Соединим связанные нуклеотиды диагоналями. Каждой вторичной структуре взаимно
однозначно соответствует набор из n непересекающихся диагоналей.
Обозначим за 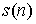 число различных
расположений n непересекающихся диагоналей в правильном число различных
расположений n непересекающихся диагоналей в правильном 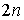 -угольнике, что отвечает рассмотрению вторичной структуры в
виде хорд на окружности с выполнением стерического условия. Тогда (с учетом
того, что вероятность комплементарности выбранных нуклеотидов равна -угольнике, что отвечает рассмотрению вторичной структуры в
виде хорд на окружности с выполнением стерического условия. Тогда (с учетом
того, что вероятность комплементарности выбранных нуклеотидов равна 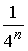 ) имеем ) имеем
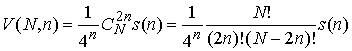 (1.2) (1.2)
Вывод формулы для s(n). Выведем формулу для функции 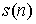 , описывающую количество способов проведения n непересекающихся диагоналей в выпуклом 2n-угольнике, включая в понятие
"диагонали" также и стороны фигуры. Действуя по индукции,
предположим , описывающую количество способов проведения n непересекающихся диагоналей в выпуклом 2n-угольнике, включая в понятие
"диагонали" также и стороны фигуры. Действуя по индукции,
предположим 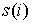 , , 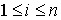 известными и примем известными и примем 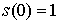 . Зафиксируем в многоугольнике вершину и проведем через нее
какую-нибудь диагональ (см. Рис. 1.9). . Зафиксируем в многоугольнике вершину и проведем через нее
какую-нибудь диагональ (см. Рис. 1.9).
Пусть диагональ является стороной 2n-угольника. Удалим из
его ломаной границы эту диагональ, пару вершин и стороны, соединяющие эти вершины
с соседними, и замкнем новую ломаную, получив многоугольник с 2(n-1) вершинами. Таким образом, зафиксировав эту диагональ, получим 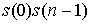 вариантов проведения
непересекающихся диагоналей. Через одну вершину многоугольника, в том числе и
через зафиксированную нами, проходят 2 стороны, и зафиксировав вторую из них,
мы также получим вариантов проведения
непересекающихся диагоналей. Через одну вершину многоугольника, в том числе и
через зафиксированную нами, проходят 2 стороны, и зафиксировав вторую из них,
мы также получим 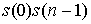 вариантов. вариантов.
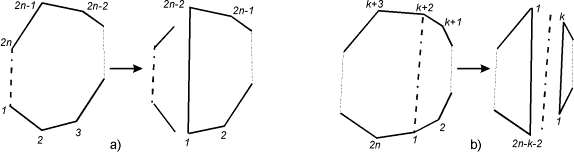
Рис. 1.9. При разбиении многоугольника
диагональю, стороны включаются в число диагоналей. a) диагональ является стороной многоугольника, в
результате разбиения получаются 0-угольник
и (2n-2)-угольник). b)
диагональ не является стороной многоугольника, в результате получаются k-угольник и (2n-k-2)-угольник).
Пусть диагональ не является стороной. Пара
вершин, соединенных этой диагональю, разобьет многоугольник на 2 ломаные, лежащие в разных полуплоскостях
относительно нее. К каждой из них применим ту же операцию удаления-замыкания,
что и в предыдущем случае, и получим 2
новых многоугольника с вершинами, количество которых будет одинаковой четности:
k и 2n - k - 2. Понятно, что в многоугольнике с нечетным
количеством вершин g можно провести не более 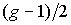 непересекающихся
диагоналей: каждая диагональ соединяет 2
вершины, и одна вершина многоугольника останется свободной. Таким образом, если
k нечетное, то в сумме в двух полученных
многоугольниках мы сможем провести не более непересекающихся
диагоналей: каждая диагональ соединяет 2
вершины, и одна вершина многоугольника останется свободной. Таким образом, если
k нечетное, то в сумме в двух полученных
многоугольниках мы сможем провести не более 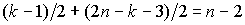 непересекающихся
диагоналей, что не соответствует нашей цели: проведению n диагоналей. Следовательно, k должно быть четным (k
= 2p). Тогда нужное нам число вариантов будет непересекающихся
диагоналей, что не соответствует нашей цели: проведению n диагоналей. Следовательно, k должно быть четным (k
= 2p). Тогда нужное нам число вариантов будет 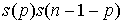 . Проводя в
исходном многоугольнике различные диагонали из фиксированной точки и суммируя
получающиеся количества вариантов, получим формулу: . Проводя в
исходном многоугольнике различные диагонали из фиксированной точки и суммируя
получающиеся количества вариантов, получим формулу:
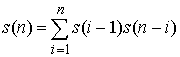 (1.3) (1.3)
Вычислим по этой формуле 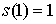 , , 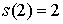 , , 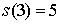 . Эти значения доказывают
базу индукции. . Эти значения доказывают
базу индукции.
Оценим функцию снизу при n > 2. В такой сумме есть 2 слагаемых вида 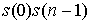 . Так как все слагаемые положительны, то . Так как все слагаемые положительны, то
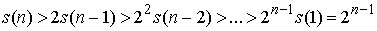 (1.4) (1.4)
При n = 2 достигается равенство.
Проведем оценку сверху. Слагаемых в сумме n, и каждое меньше, чем 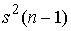 : :
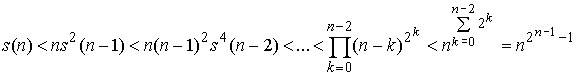 (1.5) (1.5)
При n = 2 также достигается равенство.
Полученное рекуррентное соотношение
представляет собой вариант известных чисел
Каталана 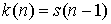 , о которых можно прочитать в [3]. Для чисел Каталана
известно и прямое выражение: , о которых можно прочитать в [3]. Для чисел Каталана
известно и прямое выражение:
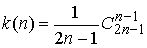 (1.6) (1.6)
Поскольку 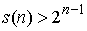 , то , то
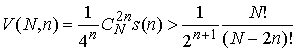 (1.7) (1.7)
причём степень полинома от N, стоящего в правой части, равна 2n. Отсюда можно сделать
два вывода:
С ростом молекулярной цепи количество
возможных вторичных структур, состоящих из n Уотсон-Криковских
связей, растет как 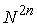 . .
С ростом молекулярной цепи количество всех
возможных вторичных структур растет быстрее, чем любой полином (иначе говоря,
скорость роста количества вторичных структур больше полиномиальной).
Для транспортных РНК средние значения
длины молекулы N=75 и количество Уотсон-Криковских связей
составляет n=20. Подставляя эти значения в формулу (1.2),
получим:
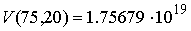
Для малых ядерных РНК, длина которых 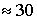 нуклеотидов, а n около 5, число возможных
структур составляет нуклеотидов, а n около 5, число возможных
структур составляет 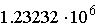 . .
В данном разделе дается строгая формулировка задачи идентификации и описываются
проблемы, возникающие при ее решении.
Отчетливо выделяются по
крайней мере 3 типа моделей пространственной структуры нуклеиновых кислот:
·
Атомарная
модель. Относится к классу задач, решаемых с использованием методов
молекулярной динамики. Молекула рассматривается как совокупность атомов
(материальных точек), которые перемещаются друг относительно друга под
воздействием межатомных сил.
·
Нуклеотидная
модель. Молекула представляется в виде цепочки элементов-нуклеотидов, связанных
между собой потенциалами фосфодиэфирных и Уотсон-Криковских связей.
·
Непрерывная
модель. В данной модели молекулярная цепь рассматривается как тонкий
непрерывный стержень с поперечными
стяжками, соответствующими Уотсон-Криковским связям.
В работе [1] описана упрощенная модель в
непрерывной постановке, где поперечные стяжки представляют собой абсолютно
твердые палочки, а стержень в свободном состоянии имеет форму винтовой линии.
При правильном выборе параметров модели она будет приближенно описывать
пространственную форму молекулы нуклеиновой кислоты. Модели такого типа уже
стали классическим средством для изучения пространственной формы молекул ДНК,
для которой экспериментальным путем удалось определить упругие параметры ее
нити. Для РНК такие параметры неизвестны.
Пространственная структура молекулы
собирается из элементов (стеблей, петель), каждый из которых представляет собой
замкнутый контур, состоящий из семейства упругих стержней, связанных жесткими
перемычками.
Выпишем систему уравнений исследуемой
модели, описывающих равновесную форму упругого стержня.
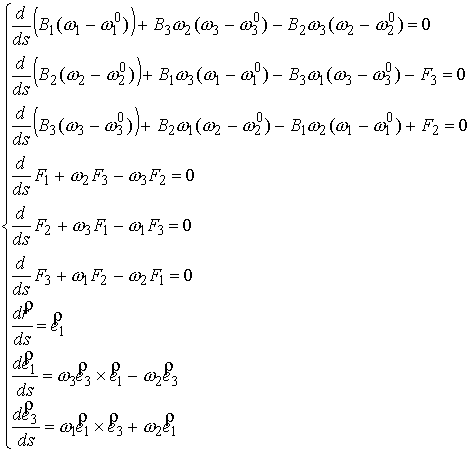 (2.1) (2.1)
Здесь 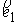 - касательный вектор
к осевой линии стержня, - касательный вектор
к осевой линии стержня, 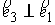 , , 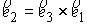 - образуют главный
сопутствующий репер осевой линии стержня, совпадающий с репером Френе при
повороте на угол - образуют главный
сопутствующий репер осевой линии стержня, совпадающий с репером Френе при
повороте на угол 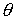 вокруг вокруг 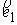 ; ; 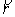 - радиус-вектор осевой линии стержня; - радиус-вектор осевой линии стержня; 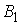 - крутильная
жесткость, - крутильная
жесткость, 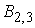 - изгибные жесткости
стержня; - изгибные жесткости
стержня; 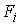 - компоненты силы,
действующей на левое поперечное сечение стержня в точке s; - компоненты силы,
действующей на левое поперечное сечение стержня в точке s; 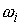 - вектор Дарбу осевой
линии стержня в изогнутом состоянии; - вектор Дарбу осевой
линии стержня в изогнутом состоянии; 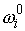 - вектор Дарбу осевой
линии стержня в свободном состоянии. - вектор Дарбу осевой
линии стержня в свободном состоянии.
Для определения пространственной формы
двуспиральных участков, где стержень в терминах модели находится в свободном
состоянии, достаточно поставить задачу Коши со следующими начальными условиями:
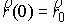 , , 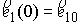 , , 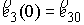 , , 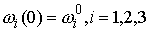 , , 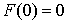
Здесь первое условие задает начальную
точку стержня, второе и третье – ориентацию главного сопутствующего репера,
четвертое и пятое фактически определяют момент и силу, приложенные к его левому
концу.
Проинтегрировав уравнения модели с
заданными начальными условиями, мы получим одну нить из двуспирального участка,
представляющую собой винтовую линию. Вторая нить получается из первой поворотом
вокруг оси винтовой линии на угол 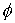 и сдвигом по этой же
оси на расстояние и сдвигом по этой же
оси на расстояние 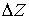 . .
Параметры данной математической модели
естественным образом разбиваются на 4 группы:
·
Упругие
параметры стержня: жесткости 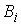 и угол и угол 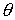 ; ;
·
Геометрические
параметры стержня для однонитевых участков, определяющие его форму в свободном
состоянии: вектор Дарбу 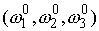 ; ;
·
3 координаты
вектора Уотсон-Криковской связи в трехграннике Френе;
·
Геометрические
параметры стержня для двуспиральных участков: вектор Дарбу 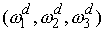 . .
Параметры 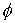 и и 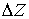 , упомянутые в предыдущем пункте, можно вычислить, используя
вектор Уотсон-Криковской связи , упомянутые в предыдущем пункте, можно вычислить, используя
вектор Уотсон-Криковской связи 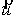 , вектор направления оси двуспирального участка , вектор направления оси двуспирального участка 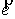 , радиус винтовой линии двуспирального участка r и вектор направления его оси , радиус винтовой линии двуспирального участка r и вектор направления его оси 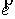 . . 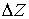 есть проекция вектора есть проекция вектора
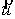 на вектор на вектор 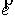 . Для определения угла . Для определения угла 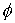 найдем проекцию
вектора найдем проекцию
вектора 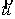 на плоскость с
нормалью на плоскость с
нормалью 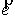 : :
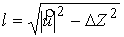 (2.2) (2.2)
и воспользуемся теоремой косинусов:
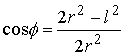 . (2.3) . (2.3)
В данной работе мы опишем
процесс идентификации 2-й группы параметров. Процесс идентификации происходит
следующим образом.
В файле, описывающем молекулу tRNA, содержатся координаты атомов, входящих в
состав молекулы, определенные путем рентгеноструктурного анализа. Из этого
множества атомов нам необходимы только координаты атомов фосфора, содержащихся
в рибозофосфатном остове молекулы, по одному на каждый нуклеотид.
Для определения геометрических параметров
стержня проще всего взять атомы фосфора из достаточно длинного стебля, так как
в терминах модели силы и моменты на концах стебля равны нулю, упругие параметры
поэтому не влияют на форму стержня и нет необходимости решать краевую задачу.
Таким образом, для нашей цели необходимо взять атомы фосфора, лежащие на одной
нити длинного стебля молекулы. После этой операции у нас появится набор точек
(или ломаная линия) в пространстве, координаты которых есть координаты атомов
фосфора (физические точки). Численное
интегрирование уравнений модели даст также набор точек (модельных точек), описывающий форму стержня. Количество модельных
точек, или звеньев модельной ломаной, будет отличаться от количества
физических. Вариацией геометрических параметров модели необходимо добиться
того, чтобы сгенерированная модельная ломаная проходила около каждой точки из
набора по возможности ближе.
В качестве алгоритма вариации параметров
был взят генетический алгоритм поиска экстремумов.
Генетические алгоритмы (ГА) -
сравнительно новый класс вычислительных алгоритмов, предназначенных для поиска
экстремумов функций. Для поиска
экстремальных значений используется эволюционный метод поиска в пространстве
решений, позаимствованный из живой природы. Рассмотрим работу типичного ГА.
Для работы ГА необходимы две вещи.
Во-первых, необходимо установить взаимно однозначное соответствие между
параметрами оптимизируемой задачи (решениями)
и битовыми строками фиксированной длины (данный этап называют
"конструированием хромосомы"). Во-вторых, требуется т.н. fitness-функция, или функционал оценки, на входе
получающий хромосому и возвращающий некоторое число - тем больше, чем
"лучше" в терминах задачи данное решение. Следует сразу же отметить,
что для алгоритма важна лишь длина хромосомы. Что в ней находится реально,
известно лишь функционалу. Имея сконструированную хромосому и функционал
оценки, можно приступать к решению задачи.
Итак, пусть задана длина хромосомы n и fitness-функция. На начальном этапе работы генерируются k случайных чисел, имеющих разрядность n бит. Они оцениваются,
и запускается классический оператор отбора – «рулетка с весами» (Рис. 2.1). Ее
отличие от обычной рулетки заключается в том, что площадь ее k секторов пропорциональна приспособленностям наших k решений. Вероятность для попадания шарика в какой-либо сектор будет тем
выше, чем больше его площадь. Запустив рулетку k раз, получим первое
поколение, приспособленность которого в среднем (по вероятности) будет выше,
чем у предыдущего. Но новых решений при этом не получается. Для генерации новых
решений используются два оператора: кроссинговер и мутация.
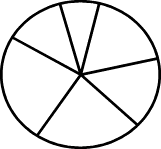
Рис. 2.1.
«Рулетка с весами». Количество секторов равно количеству особей в популяции, их
площади пропорциональны приспособленности особей.
Кроссинговер работает следующим образом.
На вход подаются две хромосомы. Случайным образом выбирается позиция (номер
бита от 1 до n), и начиная с этой позиции хромосомы меняются битами. На выходе имеются
две новые хромосомы.
Оператор мутации работает с одной
хромосомой. Случайно выбирается позиция, и бит в этой позиции инвертируется.
После кроссинговера и мутации происходит
оценка получившихся решений, затем снова запускается рулетка. Критерием
автоматической остановки является либо схождение алгоритма к одному решению
(т.е. вся популяция решений становится одинаковой), либо достижение
заданного количества итераций.
Построение хромосомы. В случае, когда у задачи имеются n параметров, для которых известны
континуумные ограниченные множества возможных значений, хромосома строится следующим
образом.
Прежде всего каждое множество
значений дискретизируется. Задаются натуральные числа 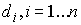 , обычно равные , обычно равные 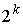 . Из i-го множества выбираются . Из i-го множества выбираются 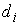 точек, обычно же
равномерно распределенные внутри множества. Затем точкам присваиваются
порядковые номера. точек, обычно же
равномерно распределенные внутри множества. Затем точкам присваиваются
порядковые номера.
Длина хромосомы задается как 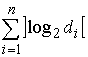 , где , где 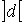 - взятие целой части от d с избытком. Внутри
хромосомы имеются n областей длины - взятие целой части от d с избытком. Внутри
хромосомы имеются n областей длины 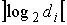 , содержащие двоичные числа, которые являются номерами точки
из соответствующего множества значений параметра. , содержащие двоичные числа, которые являются номерами точки
из соответствующего множества значений параметра.
Как было сказано выше, для работы
генетическому алгоритму необходим функционал оценки, в котором заложена
информация о решаемой задаче. Генетический алгоритм в процессе генерации
поколений будет искать его минимум. Данный функционал, получая на входе
хромосому, должен декодировать генетическую информацию, заложенную в ней,
получить численные значения параметров модели и решить задачу Коши с
полученными значениями параметров. Далее необходимо получить численную оценку
сходства пространственных форм полученной модельной ломаной и ломаной,
вершинами которой являются атомы фосфора участка реальной молекулы. Задача
оценки сходства двух ломаных осложняется тем, что, во-первых, они имеют
различное количество вершин и различные длины, во-вторых, их взаимное
расположение в пространстве с введенной системой координат далеко от
наилучшего. Таким образом, видятся два различных подхода к получению данной
оценки. Первый подход заключается в приведении двух ломаных в наилучшее
взаимное положение, при котором, например, суммарное расстояние от вершин
физической ломаной до модельной ломаной наименьшее. Второй подход состоит в
построении функционала, который не использует координатное представление ломаных
и оперирует с инвариантными относительно движений величинами. Ниже мы
рассмотрим некоторые способы построения оценочных функционалов, использующие
эти подходы.
Квазимеханический способ
сопоставления кривых. Для
приведения двух ломаных в наилучшее относительное расположение решается система
неявных разностных уравнений:
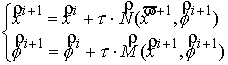 ,
(2.2) ,
(2.2)
где 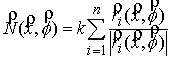 , , 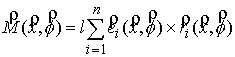 , , 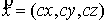 - координаты центра
масс кривой, - координаты центра
масс кривой, 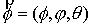 - углы поворота
кривой вокруг осей Кенига, k, l – безразмерные
коэффициенты, влияющие на скорость и характер сходимости процесса. - углы поворота
кривой вокруг осей Кенига, k, l – безразмерные
коэффициенты, влияющие на скорость и характер сходимости процесса.
На начальном этапе работы алгоритма
совмещаются центры масс кривой и набора. Затем у кривой вычисляются собственные
векторы центрального тензора инерции, и кривая поворачивается до совпадения их
с соответствующими векторами набора. Так как изначально неясно, какому вектору
набора какой вектор кривой соответствует (возможны три случая), то
рассматривают их все и выбирают тот, норма которого наименьшая.
Далее начинается итерационный процесс.
Вычисляются векторы 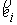 , соединяющие центр масс кривой и ближайшие точки, и векторы , соединяющие центр масс кривой и ближайшие точки, и векторы 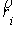 , соединяющие ближайшие точки и соответствующие им точки
набора (см. Рис. 2.2). Составляются
суммы , соединяющие ближайшие точки и соответствующие им точки
набора (см. Рис. 2.2). Составляются
суммы 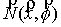 , , 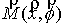 , их значения подставляются в систему неявных разностных
уравнений (2.2). , их значения подставляются в систему неявных разностных
уравнений (2.2).
Система решается методом
Эйлера. Для найденных значений координат центра масс и углов поворота вновь
вычисляются суммы 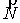 и и 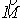 , и процесс повторяется. Критерием остановки служит
одновременная малость скалярных произведений , и процесс повторяется. Критерием остановки служит
одновременная малость скалярных произведений 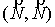 и и 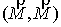 . .
Процесс минимизирует потенциал
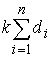 , где , где 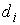 - расстояние от i-й ближайшей точки до соответствующей ей точки набора. - расстояние от i-й ближайшей точки до соответствующей ей точки набора.
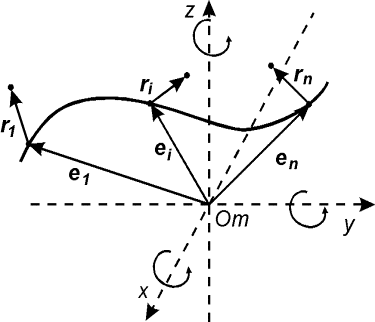
Рис. 2.2 Предварительное совмещение кривых
производится квазимеханическим методом, при котором точки 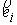 модельной кривой как
бы притягиваются к точкам модельной кривой как
бы притягиваются к точкам 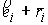 физической кривой.
Такое «притяжение» инициирует повороты вокруг координатных осей и смещение
центра масс в направлении, уменьшающем рассогласование. физической кривой.
Такое «притяжение» инициирует повороты вокруг координатных осей и смещение
центра масс в направлении, уменьшающем рассогласование.
Данный способ вычисления меры близости
пространственных форм двух ломаных имеет то преимущество, что получаемые в
конце его работы ломаные линии близки в смысле координатного их представления,
и можно, построив по координатам эти объекты, оценить визуально, насколько
хорошо форма модельной ломаной описывает форму физической. Однако, отсюда же
происходит и недостаток этого метода: непозволительно большое время расчета. Минимизируемый
потенциал имеет так называемый "овражистый рельеф". Попадая в овраги,
итерационный процесс надолго застревает в них и зачастую не доходит до
минимума.
Дискретные аналоги кривизны и кручения. Рассмотрим пространственную ломаную линию 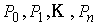 , составленную из отрезков , составленную из отрезков 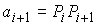 , , 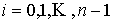 . Определим для этой ломаной две последовательности углов: . Определим для этой ломаной две последовательности углов: 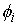 , , 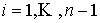 , и , и 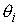 , , 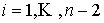 . .
Будем считать 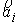 векторами с началом в
точке векторами с началом в
точке 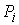 и концом в точке и концом в точке 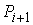 . Также примем, не ограничивая общности, что любые три
последовательно идущие вершины ломаной . Также примем, не ограничивая общности, что любые три
последовательно идущие вершины ломаной 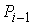 , , 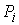 , , 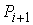 не лежат на одной
прямой. Тогда два вектора, не лежат на одной
прямой. Тогда два вектора, 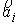 и и 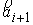 , определяют плоскость и ориентацию в ней. Определим угол , определяют плоскость и ориентацию в ней. Определим угол 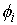 как угол между
векторами как угол между
векторами 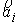 и и 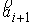 , отсчитываемый в положительном направлении согласно ориентации
плоскости. Для данного угла будут выполняться неравенства , отсчитываемый в положительном направлении согласно ориентации
плоскости. Для данного угла будут выполняться неравенства 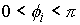 . .
Ориентированная плоскость,
построенная выше, определяет единичную нормаль 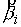 . Построим еще одну плоскость, теперь уже на векторах . Построим еще одну плоскость, теперь уже на векторах 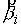 и и 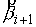 , и зададим на ней ориентацию при помощи ортогонального ей
вектора , и зададим на ней ориентацию при помощи ортогонального ей
вектора 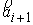 . Угол . Угол 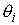 между векторами между векторами 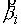 и и 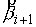 будем отсчитывать в
положительном направлении согласно этой ориентации. Тогда для этого угла будет
выполнено будем отсчитывать в
положительном направлении согласно этой ориентации. Тогда для этого угла будет
выполнено 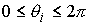 . Назовем углы . Назовем углы 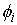 углами кривизны, а углами кривизны, а 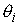 - углами кручения. - углами кручения.
Имеет место следующая теорема (доказательство см. в [2]):
Пусть заданы три
последовательности чисел: 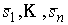 , , 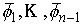 , , 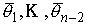 , такие, что , такие, что 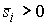 , , 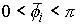 , , 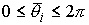 . Пусть в пространстве заданы точка . Пусть в пространстве заданы точка 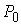 , проходящая через нее ориентированная плоскость , проходящая через нее ориентированная плоскость  и направление отрезка и направление отрезка
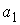 . Тогда в пространстве существует единственная ломаная с отрезками . Тогда в пространстве существует единственная ломаная с отрезками
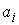 длины длины 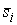 , углами кривизны , углами кривизны 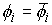 и углами кручения и углами кручения 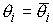 . .
Имея в виду данную теорему, можно
построить для двух ломаных 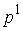 и и 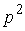 с одинаковым количеством вершин с одинаковым количеством вершин 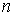 функционал с тремя
весовыми коэффициентами функционал с тремя
весовыми коэффициентами 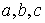 : :
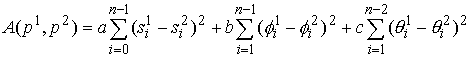 , (2.3) , (2.3)
равный 0, когда 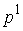 совпадает с совпадает с 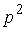 , и больший 0 в
противном случае. Но как быть, когда количество вершин у ломаных различное? , и больший 0 в
противном случае. Но как быть, когда количество вершин у ломаных различное?
Для решения этой проблемы можно применить
алгоритм редуцирования количества вершин ломаной линии.
Итак, у нас имеются две ломаные линии:
физическая, порожденная атомами фосфора из молекулы, и модельная, полученная
путем численного интегрирования уравнений равновесия. Количество вершин
модельной ломаной 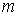 намного превышает
количество вершин намного превышает
количество вершин 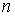 физической, к тому же
эти ломаные имеют различные длины физической, к тому же
эти ломаные имеют различные длины 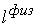 и и 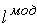 .Поделим вначале длины звеньев физической ломаной .Поделим вначале длины звеньев физической ломаной 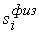 на на 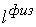 , получив относительные длины звеньев , получив относительные длины звеньев 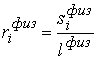 . Попробуем задать лежащие на модельной ломаной . Попробуем задать лежащие на модельной ломаной 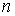 новых вершин новых вершин 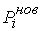 так, чтобы
выполнялись равенства так, чтобы
выполнялись равенства 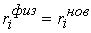 , где , где 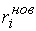 вычисляются для
ломаной с новыми вершинами вычисляются для
ломаной с новыми вершинами 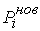 по тому же алгоритму,
что и по тому же алгоритму,
что и 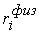 . Потребуем также, чтобы новые начальная и конечная вершины
совпадали со старыми. . Потребуем также, чтобы новые начальная и конечная вершины
совпадали со старыми.
Зафиксировав некоторое значение параметра 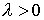 , построим сферу , построим сферу 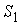 радиуса радиуса 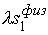 с центром в начальной точке модельной ломаной с центром в начальной точке модельной ломаной 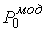 = =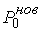 . Пусть сфера пересечет модельную ломаную, возможно, в
нескольких точках. Выберем из них ту, путь к которой от центра сферы по ломаной
меньше. Обозначим эту точку за . Пусть сфера пересечет модельную ломаную, возможно, в
нескольких точках. Выберем из них ту, путь к которой от центра сферы по ломаной
меньше. Обозначим эту точку за 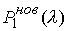 , отбросим мысленно часть ломаной, ограниченной точками , отбросим мысленно часть ломаной, ограниченной точками 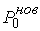 и и 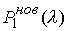 , и построим сферу , и построим сферу 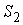 радиуса радиуса 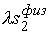 с центром в точке с центром в точке 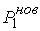 . Пусть, опять же, сфера пересечет модельную ломаную в нескольких
точках (без учета отброшенной части). Снова отберем среди них одну по принципу,
указанному выше, обозначим ее за . Пусть, опять же, сфера пересечет модельную ломаную в нескольких
точках (без учета отброшенной части). Снова отберем среди них одну по принципу,
указанному выше, обозначим ее за 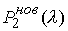 … и будем продолжать так до тех пор, пока либо не построим
все точки … и будем продолжать так до тех пор, пока либо не построим
все точки 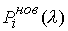 количеством количеством 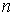 , либо не обнаружим на шаге , либо не обнаружим на шаге 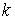 , что остаток модельной ломаной целиком лежит внутри сферы , что остаток модельной ломаной целиком лежит внутри сферы 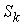 . Тогда мы проведем из точки . Тогда мы проведем из точки 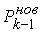 луч, направленный по
вектору луч, направленный по
вектору 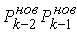 , и продолжим
процесс, двигаясь далее по лучу вместо ломаной, пока не построим все , и продолжим
процесс, двигаясь далее по лучу вместо ломаной, пока не построим все 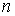 точек. точек.
После проведения данного построения можно
написать функцию
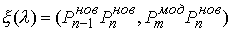 , , 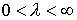 , (2.4) , (2.4)
где 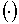 - скалярное
произведение векторов, со следующими свойствами: - скалярное
произведение векторов, со следующими свойствами:
·
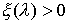 , если точка , если точка 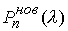 лежит вне модельной
ломаной; лежит вне модельной
ломаной;
·
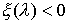 , если точка , если точка 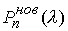 лежит внутри
модельной ломаной; лежит внутри
модельной ломаной;
·
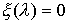 , если точка , если точка 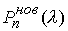 совпадает с концом
модельной ломаной. совпадает с концом
модельной ломаной.
Выберем отрезок 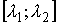 такой, что такой, что 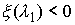 и и 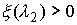 . Если для любого . Если для любого 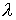 из данного
отрезка при построении точек из данного
отрезка при построении точек 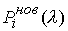 не встречаются ситуации,
подобные показанной на Рис. 2.3, то можно утверждать, что: не встречаются ситуации,
подобные показанной на Рис. 2.3, то можно утверждать, что:
·
функция 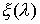 непрерывна на отрезке непрерывна на отрезке
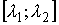 ; ;
·
ее
единственный нуль находится внутри отрезка.
Тогда нуль этой функции можно найти,
например, методом деления отрезка пополам.
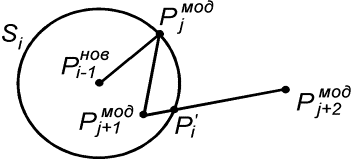
Рис. 2.3 Случай скачка функции 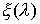 : : 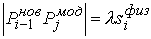 . При увеличении . При увеличении 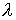 на на 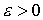 произойдет скачок
положения точки произойдет скачок
положения точки 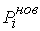 на ломаной из положения на ломаной из положения
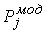 в точку в точку 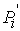 пересечения отрезка пересечения отрезка 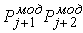 и сферы и сферы  . .
Таким образом, из ломаной 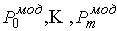 мы получили ломаную мы получили ломаную 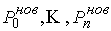 , с числом вершин, равным количеству вершин , с числом вершин, равным количеству вершин 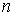 физической ломаной, и
дополнительным полезным свойством физической ломаной, и
дополнительным полезным свойством 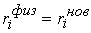 . Теперь можно подсчитать значение функционала (2.3). . Теперь можно подсчитать значение функционала (2.3).
Построенный функционал
замечателен тем, что он не зависит от взаимной ориентации ломаных и значений
координат их вершин. Он оперирует лишь с их пространственными формами. В то же
время функционал имеет и существенный недостаток: при вычислении его значения
складываются величины различных размерностей.
Построение тетраэдров. Ломаная, полученная путем интегрирования
уравнений модели, редуцируется до физической ломаной с меньшим числом звеньев
по алгоритму, описанному в предыдущем пункте. Далее в обеих ломаных проводятся
следующие построения:
·
Измеряются
расстояния между соседними точками, включая первую и последнюю (т.е. ломаная
достраивается до замкнутого многозвенника).
·
В
получившемся многозвеннике проводятся все хорды, соединяющие точки, между
которыми лежит одна вершина. Те же действия производятся для точек, между
которыми лежат две вершины.
·
Зафиксировав
длины хорд и сторон в каждом многозвеннике, мы получим неизгибаемую
конструкцию, состоящую из набора тетраэдров, построенных на вершинах
многозвенника.
Вычислив сумму квадратов разностей между
соответствующими хордами и сторонами, получим число, количественно выражающее
различие между пространственными формами двух ломаных.
Функционал данного типа, сохраняя
достоинство предыдущего - независимость от координатного представления ломаных,
свободен от его недостатка: теперь все действия совершаются над величинами
одинаковой размерности.
В качестве параметров двухнитевого участка
молекулы РНК, помимо вектора Дарбу 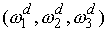 , можно указать также угол , можно указать также угол 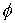 и смещение и смещение 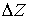 , упомянутые в разделе 2.2. Их значения можно получить,
используя знание вектора Уотсон-Криковской связи и вектора направления оси
двуспирального участка, как было указано в разделе 2.3. , упомянутые в разделе 2.2. Их значения можно получить,
используя знание вектора Уотсон-Криковской связи и вектора направления оси
двуспирального участка, как было указано в разделе 2.3.
При построении моделей однонитевых
участков молекулы РНК, каковыми являются петли различных типов, возникает
необходимость в решении краевых задач для системы уравнений (2.1). Знание
параметров Уотсон-Криковской связи необходимо для вычисления граничных условий
этих краевых задач. Однако, на практике в этих двух случаях удобнее
использовать непосредственно параметры 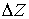 и и 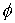 . С помощью несложных геометрических построений можно
получить из них и координаты вектора Уотсон-Криковской связи в главном сопутствующем
репере стержня. . С помощью несложных геометрических построений можно
получить из них и координаты вектора Уотсон-Криковской связи в главном сопутствующем
репере стержня.
В данном разделе описывается алгоритм,
позволяющий определить параметры 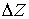 и и 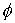 и найти вектор оси
двуспирального участка молекулы РНК. и найти вектор оси
двуспирального участка молекулы РНК.
Необходимо отметить, что
Уотсон-Криковской связью мы будем называть отрезок, соединяющий два атома
фосфора, которые принадлежат спаренным нуклеотидам молекулы.
Из рентгеноструктурных данных
о третичной структуре молекулы РНК нам известны, в частности, координаты атомов
фосфора, находящихся в двуспиральном участке молекулы. Так как модель
двуспирального участка представляет собой две винтовые линии с общей осью,
попробуем найти направление этой оси для данного участка. Для этого построим
цилиндр бесконечной длины, сумма квадратов расстояний от атомов фосфора до
поверхности которого минимальна. Его осевую линию будем считать осевой линией
двуспирального участка.
Теперь можно найти параметры 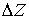 и и 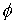 . Подсчитав среднее арифметическое проекций векторов
Уотсон-Криковских связей в двуспиральном участке на осевую линию участка, мы
получим смещение . Подсчитав среднее арифметическое проекций векторов
Уотсон-Криковских связей в двуспиральном участке на осевую линию участка, мы
получим смещение 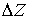 . Для вычисления угла . Для вычисления угла 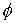 нам потребуется
среднее арифметическое длин Уотсон-Криковских связей в стебле нам потребуется
среднее арифметическое длин Уотсон-Криковских связей в стебле 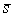 . Воспользуемся формулой (2.2), подставив в нее в качестве . Воспользуемся формулой (2.2), подставив в нее в качестве 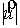 среднюю длину среднюю длину 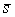 , и получим среднюю длину проекции связи на плоскость, перпендикулярную
оси цилиндра. Зная радиус цилиндра, вычислим значение , и получим среднюю длину проекции связи на плоскость, перпендикулярную
оси цилиндра. Зная радиус цилиндра, вычислим значение 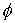 , используя формулу (2.3). , используя формулу (2.3).
Построение наилучшего цилиндра. Итак, у нас имеется множество точек 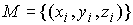 , , 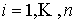 , для которого нужно построить бесконечный цилиндр таким
образом, чтобы сумма квадратов расстояний от точек множества до цилиндра была
минимальной. , для которого нужно построить бесконечный цилиндр таким
образом, чтобы сумма квадратов расстояний от точек множества до цилиндра была
минимальной.
Бесконечный цилиндр можно однозначно
задать следующим набором параметров:
·
вектор 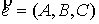 , , 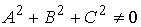 , определяющий направление оси цилиндра; , определяющий направление оси цилиндра;
·
точка 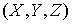 , принадлежащая оси цилиндра; , принадлежащая оси цилиндра;
·
радиус
цилиндра 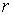 . .
Найдем выражение для суммы квадратов
расстояний от точек из множества 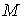 до произвольного
цилиндра. Вначале проведем плоскость до произвольного
цилиндра. Вначале проведем плоскость 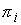 , проходящую через точку , проходящую через точку 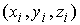 и перпендикулярную
оси цилиндра: и перпендикулярную
оси цилиндра:
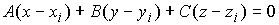 (2.5) (2.5)
Найдем координаты точки пересечения
плоскости 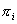 с осью цилиндра. Для
этого запишем уравнение для оси цилиндра в параметрической форме и подставим
его в (2.5): с осью цилиндра. Для
этого запишем уравнение для оси цилиндра в параметрической форме и подставим
его в (2.5):
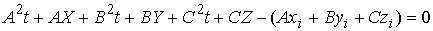
Отсюда найдем нужные нам координаты 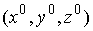 : :
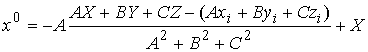 ; ;
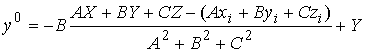 ; ;
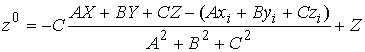 . .
Длина вектора 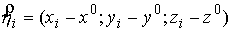 будет равна
расстоянию от будет равна
расстоянию от 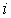 -й точки множества -й точки множества 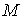 до оси цилиндра.
Тогда сумма квадратов расстояний от точек множества до цилиндра будет выглядеть
следующим образом: до оси цилиндра.
Тогда сумма квадратов расстояний от точек множества до цилиндра будет выглядеть
следующим образом:
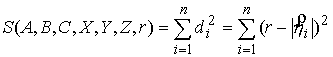
Аналитически из условия 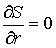 можно найти
оптимальное значение радиуса цилиндра: можно найти
оптимальное значение радиуса цилиндра:
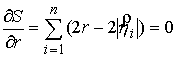 , ,
откуда 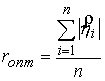 , т.е. оптимальный радиус равен среднему арифметическому
расстояний от точек из множества , т.е. оптимальный радиус равен среднему арифметическому
расстояний от точек из множества 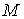 до оси цилиндра.
Точное нахождение оптимальных значений других параметров цилиндра представляет
уже серьезную проблему. Задачу поиска минимума функции до оси цилиндра.
Точное нахождение оптимальных значений других параметров цилиндра представляет
уже серьезную проблему. Задачу поиска минимума функции 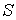 можно решить
численно. Для этого желательно еще больше уменьшить количество параметров. можно решить
численно. Для этого желательно еще больше уменьшить количество параметров.
Не ограничивая общности, можно наложить на
вектор оси цилиндра 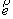 условие условие 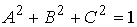 . При этом вектор . При этом вектор 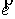 будет определяться
уже двумя параметрами. Попробуем найти эту зависимость, предполагая областью
допустимых значений двух параметров a и b всю числовую прямую. будет определяться
уже двумя параметрами. Попробуем найти эту зависимость, предполагая областью
допустимых значений двух параметров a и b всю числовую прямую.
Введем в трехмерном пространстве декартову
систему координат. Вокруг точки O построим сферу радиуса 1. Через две точки, 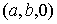 и и 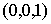 , проведем прямую, которая будет иметь со сферой две точки
пересечения: , проведем прямую, которая будет иметь со сферой две точки
пересечения: 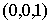 и и 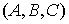 . Нужное свойство, . Нужное свойство, 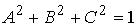 , следует из способа построения точки. Выпишем выражения для A, B и C через значения параметров a и b: , следует из способа построения точки. Выпишем выражения для A, B и C через значения параметров a и b:
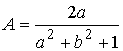 , , 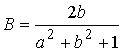 , , 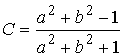 . .
При данном способе
параметризации можно получить все возможные векторы 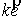 , , 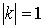 , с точностью до знака коэффициента k, знание которого не нужно для задания направления оси цилиндра. Выражения
для A, B и C являются непрерывными
функциями от a и b. , с точностью до знака коэффициента k, знание которого не нужно для задания направления оси цилиндра. Выражения
для A, B и C являются непрерывными
функциями от a и b.
Для задания точки, лежащей на
оси цилиндра, также достаточно двух параметров x и y. Эти параметры представляют собой координаты на плоскости,
перпендикулярной вектору 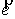 . Зависимость трехмерных координат . Зависимость трехмерных координат 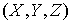 этой точки от x и y можно найти следующим образом. этой точки от x и y можно найти следующим образом.
Если вектор  коллинеарен базисному
вектору коллинеарен базисному
вектору 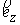 , то значения трехмерных координат получаются автоматически: , то значения трехмерных координат получаются автоматически: 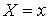 , , 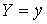 , , 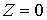 . В противном случае найдем вектор . В противном случае найдем вектор 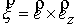 и угол между
единичными векторами и угол между
единичными векторами 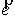 и и 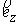 : :
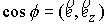 , , 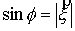 , ,
и запишем выражение:
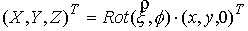
Здесь 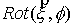 - матрица - матрица  , реализующая поворот вокруг вектора , реализующая поворот вокруг вектора 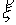 с началом в точке O на угол с началом в точке O на угол 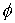 ([4]). ([4]).
Таким образом, мы свели число параметров
задачи до четырех, что значительно ускорило нахождение минимума функции 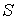 методом градиентного
спуска. методом градиентного
спуска.
Заметим, что функция расстояния непрерывно
зависит от параметров цилиндра (ориентации и положения оси, радиуса).
Пространство ориентации оси цилиндра компактно (RP2). При неограниченном сдвиге оси, а также увеличения радиуса, функция
расстояния неограниченно возрастает. Отсюда следует существование минимума у
функции расстояния, т.е. минимальный цилиндр всегда существует. Однако единственность
минимального цилиндра не гарантируется.
Вычисление граничных условий на примере шпилечной петли. Для нахождения формы шпилечной петли
необходимо вычислить значения следующих величин:
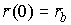 , , 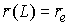 , , 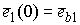 , , 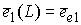 , , 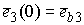 , , 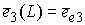 . .
Здесь 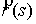 -радиус-вектор, -радиус-вектор, 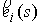 - главный сопутствующий репер осевой линии стержня,
совпадающий с репером Френе при повороте на угол - главный сопутствующий репер осевой линии стержня,
совпадающий с репером Френе при повороте на угол 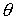 вокруг вокруг 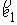 . .
На Рис. 1.4 b) видно, что шпилечная петля находится на
конце двуспирального участка молекулы. Следовательно, относительное
расположение точек 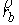 и и 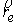 должно совпадать с расположением
концов двуспирального участка, а должно совпадать с расположением
концов двуспирального участка, а 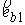 , , 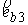 , , 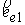 и и 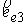 задаются положениями
главных сопутствующих реперов на концах стебля ([1]). задаются положениями
главных сопутствующих реперов на концах стебля ([1]).
Пусть осевая линия
двухспирального участка совпадает с осью Oz. Зададим координаты
точки 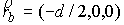 . Главный сопутствующий репер в этой точке (вектора . Главный сопутствующий репер в этой точке (вектора 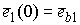 и и 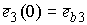 ) нам известен из
решения задачи Коши для одной нити двуспирального участка. Необходимо найти
репер и положение конца второй нити. Для этого применим к точке ) нам известен из
решения задачи Коши для одной нити двуспирального участка. Необходимо найти
репер и положение конца второй нити. Для этого применим к точке 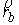 и векторам и векторам 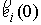 операцию поворота
вокруг оси Oz на угол операцию поворота
вокруг оси Oz на угол 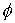 , а затем операцию параллельного переноса на вектор , а затем операцию параллельного переноса на вектор 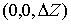 . В результате этих действий мы получим значения координат
точки . В результате этих действий мы получим значения координат
точки 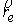 и векторов и векторов 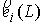 . .
Решение краевой задачи необходимо при
проведении второго этапа идентификации – определения упругих параметров стержня.
В данном разделе приводятся результаты
некоторых вычислительных экспериментов по оценке геометрических параметров
двуспиральных участков РНК на основе рентгеноструктурных данных.
При использовании генетических алгоритмов
поиска экстремумов очень важным является правильный выбор параметров этого
алгоритма. От этого зависит быстродействие процесса, скорость его сходимости,
точность окончательного результата. Нами были опробованы различные вектора
параметров. Наиболее сбалансированным с точки зрения указанных требований
оказался следующий вектор параметров:
Число идентифицируемых параметров: 2 (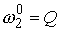 , , 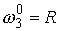 ) )
Число особей в популяции: 70
Количество бит на параметр: 8
Вероятность кроссинговера: 1.0
Вероятность мутации: 0.08
Оператор кроссинговера:
двухточечный
Оператор мутации: замена
каждого бита на случайный с вероятностью мутации
Оператор селекции: турнирный.
В качестве алгоритма сравнения двух
ломаных был взят квазимеханический алгоритм. В процессе работы генетического
алгоритма минимизировалось максимальное расстояние между кривой и набором
атомов фосфора.
Следует сказать несколько слов о
корректности поиска экстремумов в данной задаче. Конечно, в задаче
сопоставления геометрических форм пространственных кривых функционал сравнения
может иметь не одну точку зкстремума. Следует ожидать, что их может быть
достаточно много. Поэтому в строгом математическом смысле задача не является
корректной (т. е. ее решение не единственно). Однако, наличие минимума у
оценочного функционала можно гарантировать, т. к. он ограничен снизу на
компактном пространстве параметров. Поэтому осмысленной является задача
изменения геометрических параметров модели таким образом, чтобы оценочный
функционал стал меньше при разумной форме и взаимном расположении
сопоставляемых кривых в точке зкстремума. В силу сказанного результаты процесса
идентификации контролировались нами для того, чтобы отбрасывать заведомо
неподходящие варианты.
Ниже приводятся результаты наиболее
удачных процессов идентификации параметров.
Для идентификации параметров выбран
участок нити из двухнитевого стебля молекулы tRNA-Phe SACCHAROMYCES CEREVISIAE ([14]), состоящая из пяти нуклеотидов. Идентифицировались параметры Q и R, изменяющиеся в пределах 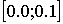 . Для достижения
минимума функционала генетический алгоритм сгенерировал порядка 300 поколений
за время 2 часа (на машине класса Pentium166-MMX). . Для достижения
минимума функционала генетический алгоритм сгенерировал порядка 300 поколений
за время 2 часа (на машине класса Pentium166-MMX).
В результате идентификации для параметров
были найдены следующие значения:
Q = 0.08981 R = 0.05098 func = 0.11727
Визуальный результат
идентификации представлен на Рис. 3.1.
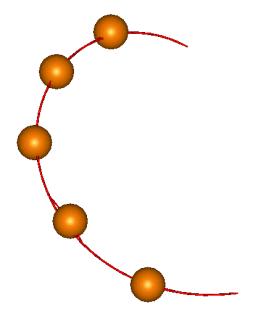
Рис. 3.1. Упругий стержень,
аппроксимирующий участок из 5 нуклеотидов. Шарики соответствуют атомам фосфора,
положение которых определено методом рентгеноструктурного анализа. Непрерывная
линия соответствует нити двухспирального участка, полученной в результате
процесса идентификации.
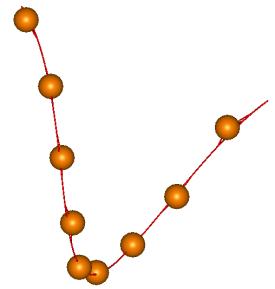
Рис. 3.2.
Упругий стержень, аппроксимирующий участок из 9 нуклеотидов. Смысл изображения
такой же, как на Рис. 3.1.
Из двухнитевого стебля той же
молекулы был выбран участок, состоящий из девяти нуклеотидов. Для параметров Q и R и области их изменения 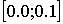 минимум был найден спустя 90 поколений,
сгенерированных за 4 часа. Значения параметров получились такие: минимум был найден спустя 90 поколений,
сгенерированных за 4 часа. Значения параметров получились такие:
Q = 0.0845 R = 0.0375 func = 0.8072744
Представление о том, как
выглядит форма стержня, можно получить из Рис. 3.2.
1. Козлов Н. Н, Кугушев Е. И., Сабитов Д. И.,
Энеев Т. М. Компьютерный анализ процессов структурообразования нуклеиновых
кислот. Препринт ИПМ им М. В. Келдыша РАН, 2002, N 42.
2. Аминов Ю. А. Дифференциальная геометрия и
топология кривых. Наука, 1987.
3. Ширшов А. Об одной комбинаторной задаче.
«Квант» № 9, 1979, с. 19
4. Бронштейн И. Н., Семендяев К. А.
Справочник по математике для инженеров и учащихся втузов. - 13-е изд.,
исправленное. Наука, 1986.
5. Батищев Д. И. Генетические алгоритмы
решения экстремальных задач / Под ред. академика АЕН Львовича Я. Е.: Учеб.
пособие. Воронеж. гос. техн. ун-т; Нижегородский гос. ун-т. Воронеж, 1995.
6. Энеев Т.М., Козлов Н.Н.,
Кугушев Е.И. Процессы структуризации биомолекул. Результаты
математического моделирования. Препринт
ИПМ им. М.В. Келдыша РАН, N 69, 1995, с. 22.
7. Козлов Н.Н., Кугушев Е.И.,
Энеев Т.М. Структурообразующие характеристики
транскрипционного процесса. Математическое моделирование т.10, N 6, с.3-19, 1998.
8. www.moldyn.ru/library/md/default.htm
9. Tomb et al. The complete genome sequence of
the gastric pathogen Helicobacter pyori. Nature 1997, Aug 7; 388 (6642).
10. Brown R. S., Dewan J. C., Klug A.
Crystallographic and biochemical investigation of the lead-catalyzed hydrolysis
of yeast phenilalanine tRNA. Biochemistry. v. 24, 1985.
11. http://www.tulane.edu/~biochem/nolan/lectures/rna/rnapro02.htm
12. http://wwwmgs.bionet.nsc.ru/mgs/gnw/selex
13. Попов
Е.П. (1948) Нелинейные задачи статики
тонких стержней. Л.М. ОГИЗ, с.170.
14. Benham C.J. (1983) Geometry and mechanics of
DNA superhelicity. Biopolymers, 22,
11, 2477 - 2495.
|